“©Έο δΥΆΗ≈ΡνΒΡ÷ς“ΣΧτ’Ϋ‘Ύ”ΎΫΪΉψΙΜΦΝΝΩΒΡœΗΑϊΕΨ–‘“©Έο δΥΆΒΫΧΊΕ®ΈΜ÷ΟΘ§Ά§ ±ΫΪ≤ΜΝΦΗ±Ής”ΟΫΒ÷ΝΉνΒΆΓΘΈΣΩΥΖΰ’β“ΜΡ―ΧβΘ§ΡΩ«Α’ΐ≤…”ΟΕύ÷÷ΖΫΖ®ΓΘ’β–©ΖΫΖ®Αϋά®ΒΪ≤Μœό”ΎΘΚaΘ©άϊ”Ο“©Έο δΥΆ‘ΊΧεΚΆ÷ΤΦΝΘ§»γΡ…ΟΉΝΘΉ”ΓΔ±≠ΖΦΧΰΜρΜΖΚΐΨΪΘ§ΫΪœΗΑϊΕΨ–‘“©ΈοΉΑ‘ΊΤδ÷–Θ§≤Δ‘ΎΕώ–‘÷ΉΝω≤ΩΈΜ ΆΖ≈ΘΜbΘ©‘Ύ÷ΉΝωΈΔΜΖΨ≥Θ®»γΒΆ pH ÷ΒΘ©÷–Α≤ΉΑ≤ΜΈ»Ε®ΒΡΜ·―ßΜυΆ≈Θ§“‘―ΎΗ«œΗΑϊΕΨ–‘“©ΈοΘ§–Έ≥…ΨΏ”–‘ω«ΩΒΡ―ΣΫ§Έ»Ε®–‘ΚΆ/ΜρœΗΑϊ…χΆΗ–‘ΒΡ«Α“©Θ§Ά§ ±ΫΒΒΆΕ‘’ΐ≥ΘœΗΑϊΒΡΕΨ–‘ΘΜcΘ©ΫΪ“©ΈοΙ≤ΦέΝ§Ϋ”ΒΫ÷ΉΝωΑ–œρ‘ΣΦΰΘ®–ΓΖ÷Ή”ΓΔκΡΜρΩΙΧεΘ©…œΘ§ ΙΤδΡήΙΜ―Γ‘ώ–‘ΒΊΑ–œρ≤Δ…χΆΗΑ©œΗΑϊΓΘ’β÷÷Ν§Ϋ” «Ά®ΙΐΚœάμ…ηΦΤΒΡΝ§Ϋ”Χε Βœ÷ΒΡΘ§ΗΟΝ§Ϋ”ΧεΡήΙΜ‘ΎΑ©œΗΑϊΈΔΜΖΨ≥÷– ΆΖ≈“©Έο
άμœκΒΡΑ–œρΖ÷Ή”ΉΑ÷Ο”ΠΑϋΚ§“‘œ¬ΡΘΩιΘΚaΘ©œΗΑϊΕΨ–‘“©ΈοΘ®“©ΈοΘ©Θ§bΘ©“©Έο‘Υ δ‘ΊΧεΘ®άΐ»γ÷§÷ ΓΔΗ ¬ΕΨέΧ«Θ©Θ§cΘ©ΫΪ‘Υ δ‘ΊΧε”κœΗΑϊΕΨ–‘Β·ΆΖΝ§Ϋ”ΒΡΝ§Ϋ”ΧεΘ§dΘ©ΓΑΩ…±ύ≥ΧΓ±ΒΡΒΦΚΫ/Α–œρ≤ΩΖ÷Θ®άΐ»γ ήΧεΧΊ“λ–‘≈δΧεΘ©Θ§“‘ΦΑ eΘ©ΓΑ“ΰ…μΓ±‘ΊΧεΘ®άΐ»γΨέ““Εΰ¥ΦΘ©“‘ΧαΗΏ…ζΈοάϊ”ΟΕ»ΓΘ’β–©ΡΘΩι‘ΎΆΦ 1A ÷–“‘≤ΜΆ§―’…Ϊ±ύ¬κΘΚ‘Υ δ‘ΊΧεΈΣ¬Χ…ΪΘ§“©ΈοΈΣάΕ…ΪΘ§Ν§Ϋ”ΧεΈΣΚλ…ΪΘ§ΒΦΚΫ/Α–œρ≤ΩΖ÷ΈΣΚΎ…ΪΘ§ΓΑ“ΰ…μΓ±‘ΊΧεΈΣΜ“…ΪΓΘ
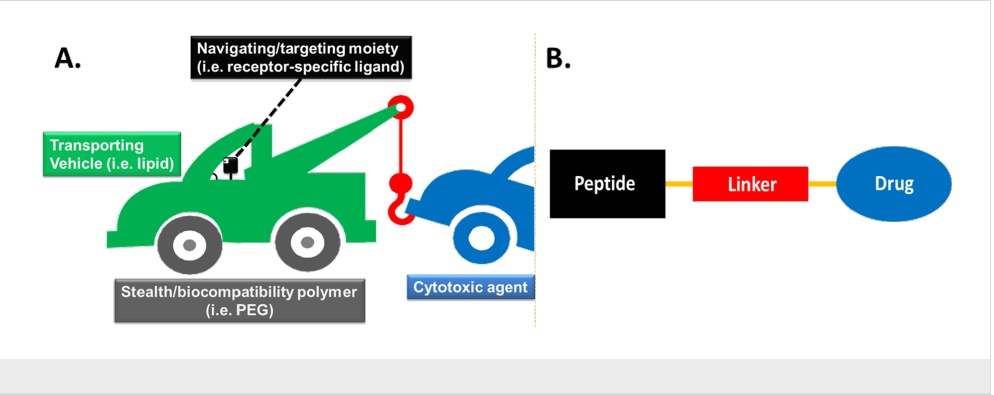
ΆΦ1 άμœκΒΡΒΦΚΫ“©Έο δΥΆœΒΆ≥ΒΡΉήΧεΫαΙΙΦήΙΙΓΘB. κΡ-“©Έο≈ΦΝΣΈοΘ®PDCΘ©ΒΡ“ΜΑψΫαΙΙΓΘ
ΕύκΡ“©Έο≈ΦΝΣΈοΘ®PDCΘ©ΒΡ÷ΈΝΤ–ßΙϊ÷ς“Σ»ΓΨω”Ύ“©ΈοΒΡ–ßΝΠ“‘ΦΑΥυΉιΉΑ≈ΦΝΣΈοΒΡΑ–œρ–߬ ΓΘ“ρ¥ΥΘ§ΕύκΡ“©Έο≈ΦΝΣΈο”ΠΨΏ±ΗΡ≥–©ΧΊ–‘Θ§ ΙΤδ≥…ΈΣ”–Έϋ“ΐΝΠΒΡ÷ΈΝΤΚρ―Γ“©ΈοΘΚ
1. PDC ÷–ΥυΚ§ΒΡκΡ±Ί–κ―Γ‘ώ–‘ΒΊ«““‘ΉνΦ―«ΉΚΆΝΠ”κΧΊΕ® ήΧεΫαΚœΘ§ΗΟ ήΧε¥φ‘Ύ”ΎΑ–Ήι÷·œΗΑϊΒΡ±μΟφΘ§ΕχΖ«œΗΑϊ÷ ΜρœΗΑϊΚΥΡΎΘ®Φ¥άύΙΧ¥Φ ήΧεΘ©ΓΘ
2.Υυ―Γ ήΧε”Π‘ΎΑ©œΗΑϊ…œΧΊ“λ–‘±μ¥οΜρΙΐΕ»±μ¥οΘ®Ά®≥Θ”κ’ΐ≥ΘœΗΑϊœύ±»ΗΏ≥ω 3 ±ΕΜρΗϋΗΏΘ©ΓΘ¥ΥΆβΘ§Τδ±μ¥οΥ°ΤΫ”ΠΉψ“‘ΫΪ”––ßΦΝΝΩΒΡ“©Έο±Ο»κœΗΑϊΡΎ≤ΩΓΘ
3.κΡ‘ΊΧε”Π“‘’β―υΒΡΖΫ ΫΙΙΫ®ΘΚΡήΙΜ”κ“©ΈοΚΆ/Μρ”ΪΙβΆ≈Ϋχ––≈ΦΝΣΓΘ≈ΦΝΣΆ®≥ΘΖΔ…ζ‘ΎάΒΑ±ΥαΓΔΑκκΉΑ±ΥαΚΆΙ»Α±Υα…œΘ§Ά®Ιΐ’ΐΫΜ≈ΦΝΣΜρ‘ΎΙΧœύκΡΚœ≥…Ιΐ≥Χ÷–ΖΔ…ζ‘ΎκΡΒΡ”Έάκ N ΕΥΓΘ≤ΜΙΐΘ§≈ΦΝΣΈΜΒψ”ΠΫς…ς―Γ‘ώΘ§“ρΈΣκΡΫαΙΙΈΔΜΖΨ≥÷–ΒΡ»≈Ε·Ω…ΡήΜαΒΦ÷¬Τδ”κΑ– ήΧεΒΡΫαΚœ«ΉΚΆΝΠ/―Γ‘ώ–‘…Ξ ßΓΘ
4.Ν§Ϋ”Ή”ΒΡ―Γ‘ώ”ΠΫς…ςΘ§“‘»Ζ±ΘκΡ≈ΦΝΣ“©ΈοΘ®PDCΘ©ΖΔΜ”ΉνΦ―–‘ΡήΓΘ―Γ‘ώ≤ΜΒ±Ω…ΡήΜαΒΦ÷¬κΡ”κ ήΧεΒΡΫαΚœ«ΉΚΆΝΠΫΒΒΆΘ§“‘ΦΑ“©Έο÷ΈΝΤ¥ΑΥθ–ΓΓΘ¥ΥΆβΘ§Ν§Ϋ”Ή”‘Ύ―Σ“Κ―≠ΜΖΙΐ≥Χ÷–”ΠΨΏ”–ΟΗΈ»Ε®–‘Θ§“‘±ψ”––ßΒΫ¥οΕώ–‘÷ΉΝω≤ΩΈΜΘ§≤Δ‘ΎΤδΈΔΜΖΨ≥÷– ΆΖ≈”––ß‘ΊΚ…Θ§¥”ΕχΫΒΒΆΆ―Α–ΕΨ–‘ΓΘ
5.œΗΑϊΕΨ–‘“©Έο”ΠΚ§”–Ρή”κ÷ΉΝωΙι≥≤κΡΝ§Ϋ”ΒΡ Β±ΙΌΡήΆ≈Θ®άΐ»γΦΣΈςΥϊ±θΘ©Θ§»τ≤Μ¥φ‘Ύ‘ρ”ΠΚœάμΧμΦ”Θ§Ά§ ±ΩΦ¬«œΗΑϊΕΨ–‘“©ΈοΒΡΉν÷’―ή…ζΈοΘ§“‘±Θ≥÷Τδ‘≠”–ΒΡœΗΑϊΕΨ–‘Μν–‘ΓΘ
“‘œ¬ΗςΫΎΉήΫαΝΥ―Γ‘ώ–‘Α–œρΕώ–‘œΗΑϊΒΡκΡ-“©Έο≈ΦΝΣΈοΒΡΜυ±Ψ…ηΦΤ‘≠‘ρΓΘ
―Γ‘ώΚœ ΒΡΑ–œρκΡΘΚ
κΡάύΈο÷ Θ®œΏ–‘ΜρΜΖΉ¥Θ©÷÷άύΖ±ΕύΘ§“―±Μ”ΟΉς‘ΊΧε/Α–œρ‘ΣΥΊΘ§≥…ΙΠΒΊΫΪœΗΑϊΕΨ–‘Β·ΆΖΒίΥΆ÷ΝΑ©œΗΑϊΓΘ’β–©κΡάύΈο÷ ΨΏ”–œΗΑϊΧΊ“λ–‘Θ§Ρή”κΧΊΕ® ήΧεΫαΚœΘ§¥ΌΫχΤδΡΎΜ·ΓΘΥϋΟ«Ά®≥ΘΆ®ΙΐΑϊΆΧΉς”ΟΫχ»κœΗΑϊΘ§»ΜΚσ±Μ‘Υ δΒΫΟΗ≈®Ε»ΗϋΗΏΓΔpH ÷ΒΗϋΒΆΒΡœΗΑϊΡΎ«χ “Θ§‘ΎΡ«άοΥϋΟ«”κ ήΧεΖ÷άκΘ§ΥφΚσ”κΩΙΑ©“©ΈοΖ÷άκΓΘ”Ο”Ύ PDC ΒΡ¥ζ±μ–‘κΡάύΈο÷ Ψάΐ»γœ¬ΓΘ
RGD
“Μ÷÷ΙψΖΚ Ι”ΟΒΡκΡ‘ΊΧε «»ΐκΡΨΪΑ±Υα - Η Α±Υα - ΧλΕ§Α±ΥαΘ®RGDΘ©Μυ–ρΘ§ΗΟΜυ–ρ”Ύ 20 άΦΆ 80 Ρξ¥ζ≥θ”… Ruoslahti ΚΆ Pierschbacher ‘ΎœΥΝ§ΒΑΑΉ÷– Ή¥ΈΖΔœ÷Θ§œΥΝ§ΒΑΑΉΫιΒΦœΗΑϊΗΫΉ≈Θ§«““―÷ΣΤδΑ–œρ’ϊΚœΥΊΠΝ5Π¬1Θ§”…”Ύ÷ΉΝωΒΡΖΔ…ζΗΏΕ»“άάΒ”Ύ«®“ΤΓΔ«÷œ°ΚΆ―ΣΙή…ζ≥…Θ§“ρ¥Υ’ϊΚœΥΊ≥…ΈΣ÷Ί“ΣΒΡΩΙΑ©Α–ΒψΓΘ’ϊΚœΥΊΠΝvΠ¬3 «÷ΉΝω―ΣΙή…ζ≥…ΚΆΉΣ“ΤΒΡ÷Ί“Σ“ρΥΊΘ§’β «Α©÷Δ«χ±π”ΎΤδΥϊΦ≤≤ΓΒΡΝΫΗω≥ΘΦϊΧΊ’ςΓΘ÷ΒΒΟΉΔ“βΒΡ «Θ§‘ΎœΗΑϊ‘ω÷≥ΓΔ«÷œ°ΚΆ―ΣΙή…ζ≥…ΖΫΟφΘ§’ϊΚœΥΊΠΝvΠ¬3Θ®“≤≥ΤΈΣœΥΝ§ΒΑΑΉ ήΧεΘ©‘ΎΥυ”–’ϊΚœΥΊ÷–œ‘ΒΟΉνΈΣ÷Ί“ΣΓΘ’β÷÷’ϊΚœΥΊ‘ΎΜνΜ·ΒΡΡΎΤΛœΗΑϊΓΔ–¬…ζ―ΣΙήΚΆΤδΥϊ÷ΉΝωœΗΑϊ÷–ΙΐΕ»±μ¥οΘ§ΒΪ‘Ύ¥σΕύ ΐ≥…Ρξ…œΤΛœΗΑϊ÷–±μ¥οΥ°ΤΫΦΪΒΆΘ§ ΙΤδ≥…ΈΣΩΙ―ΣΙή…ζ≥…÷ΈΝΤΒΡάμœκΑ–ΒψΓΘ”…”ΎΤδ‘ΎΑ©œΗΑϊ÷–ΒΡΗΏ±μ¥οΥ°ΤΫΘ§Κ§RGDΜυ–ρΒΡΦΗ÷÷κΡ“―±Μ”Ο”Ύ÷Τ±ΗPDCsΘ§Τδ÷–ΉνΨΏ¥ζ±μ–‘ΒΡάΐΉ” «κΡCDCRGDCFCΓΘ
¥Ό–‘œΌΦΛΥΊ ΆΖ≈ΦΛΥΊΘ®GnRHΘ©
¥Ό–‘œΌΦΛΥΊ ΆΖ≈ΦΛΥΊΘ®GnRHΘ©Θ§“≤≥ΤΈΣ¥ΌΜΤΧε…ζ≥…ΥΊ ΆΖ≈ΦΛΥΊΘ®LHRHΘ©Θ§ «“Μ÷÷ΗΚ‘π¥”Ρ‘¥ΙΧε«Α“ΕΖ÷ΟΎΝΫ÷÷¥Ό–‘œΌΦΛΥΊΓΣΓΣ¬―≈ί¥ΧΦΛΥΊΘ®FSHΘ©ΚΆΜΤΧε…ζ≥…ΥΊΘ®LHΘ©ΒΡΦΛΥΊΓΘGnRH ”…œ¬«πΡ‘ΡΎΒΡ GnRH …ώΨ≠‘ΣΚœ≥…≤Δ ΆΖ≈Θ§―Γ‘ώ–‘ΒΊ”κΤδ ήΧεΘ®GnRH-RΘ©ΫαΚœΘ§ΗΟ ήΧε «“Μ÷÷ΤΏΩγΡΛ G ΒΑΑΉ≈ΦΝΣ ήΧεΓΘGnRH ΦΛΥΊΒΡΫαΙΙΘ®pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2Θ©”Ύ 1971 Ρξ”… Baba Β»»Υ Ή¥ΈΖΔœ÷ΓΘ≥ΐΝΥ’β÷÷–Έ ΫΆβΘ§‘Ύ¥σΕύ ΐΦΙΉΒΕ·Έο“‘ΦΑ»ΥάύΧεΡΎΜΙΖΔœ÷ΝΥ GnRH-IIΘ®pGlu-His-Trp-Ser-His-Gly-Trp-Tyr-Pro-Gly-NH2Θ©ΓΘ’β÷÷κΡΆ®Ιΐ“Μ÷÷άύΥΤΒΡ ήΧεΘ®II –Ά GnRH-RΘ©ΤπΉς”ΟΘ§ΗΟ ήΧε‘ΎΑϋά®÷ΉΝωœΗΑϊ‘ΎΡΎΒΡ≤ΜΆ§Ήι÷·÷–Ψυ”–±μ¥οΓΘΝμ“Μ÷÷Χλ»ΜΒΡ GnRH Ά§÷÷–Ά « GnRH-IIIΘ®pGlu-His-Trp-Ser-His-Asp-Trp-Lys-Pro-Gly-NH2Θ©Θ§Υϋ «¥”ΚΘΤΏ»ζς©÷–Ζ÷άκ≥ωά¥ΒΡΓΘGnRH-III ”κΑ©œΗΑϊ±μΟφΙΐ±μ¥οΒΡ GnRH-R ΫαΚœΘ§¥”Εχ≤ζ…ζΩΙ‘ω÷≥Ής”ΟΘ§ΒΪ‘Ύ¥ΧΦΛ¥ΙΧε ΆΖ≈¥Ό–‘œΌΦΛΥΊΖΫΟφΥΤΚθ≤Μ»γΤδΥϊ GnRH άύΥΤΈο”––ßΓΘ
¥Ό–‘œΌΦΛΥΊ ΆΖ≈ΦΛΥΊΘ®GnRHΘ©κΡάύΥΤΈοΙΙ≥…ΝΥ“Μάύ–¬–ΥΒΡ÷ΉΝωΙι≥≤κΡΘ§”Ο”Ύ’κΕ‘±μ¥ο GnRH ήΧεΘ®GnRH-RΘ©ΒΡΕώ–‘Ήι÷·ΓΘΤδΩΣΖΔΜυ”Ύ’β―υ“ΜΗω ¬ ΒΘ§Φ¥ΧΊΕ®ΒΡ»ΥάύΑ©œΗΑϊΘ®÷ς“Σ «¬―≥≤Α©ΓΔ«ΑΝ–œΌΑ©ΓΔΖΈΑ©ΚΆ»ιœΌΑ©Θ©œύΕ‘”Ύ’ΐ≥ΘΉι÷·Εχ―‘Θ§ΜαΧΊ“λ–‘±μ¥οΜρΙΐΕ»±μ¥ο GnRH-RΓΘΤδ÷–Ήν≥Θ”ΟΒΡ¥Ό–‘œΌΦΛΥΊ ΆΖ≈ΦΛΥΊΘ®GnRHΘ©άύΥΤΈο « D-Lys6-GnRH-IΘ§ΥϋΡή―Γ‘ώ–‘ΒΊ”κ GnRH ήΧεΘ®GnRH-RΘ©ΫαΚœΓΘΫΪΧλ»ΜΦΛΥΊ÷–ΒΡΒΎ 6 ΈΜΗ Α±ΥαΧφΜΜΈΣ D-Lys6Θ§ΒΟΒΫΝΥ“Μ÷÷ΫαΚœ«ΉΚΆΝΠΗϋΗΏΓΔΠ¬ ΉΣΫ«ΗϋΈ»Ε®«“ΡήΒ÷ΩΙΒΑΑΉΥ°ΫβΒΡάύΥΤΈοΓΘ¥ΥΆβΘ§άΒΑ±ΥαΒΡ≤ύΝ¥Κ§”–“ΜΗω”ΈάκΒΡΑ±ΜυΘ®Π≈NH₂Θ©Θ§’β ΙΒΟΥϋΡήΙΜ”κœΗΑϊΕΨ–‘Β·ΆΖΫχ––’ΐΫΜ≈ΦΝΣΓΘΜυ”Ύ GnRH ΒΡκΡ“©Έο≈ΦΝΣΈοΘ®PDCΘ© ΐΝΩ÷ΎΕύΓΘ
…ζ≥Λ“÷ΥΊΘ®SSTΘ©
…ζ≥Λ“÷ΥΊ «“Μ÷÷”……ώΨ≠ΡΎΖ÷ΟΎΓΔ―Ή÷ΔΚΆΟβ“ΏœΗΑϊ≤ζ…ζΒΡ…ώΨ≠κΡΘ§‘ΎΕύ÷÷…ζάμΙΠΡή÷–ΖΔΜ”Ή≈÷Ί“ΣΉς”ΟΘ§Ω…ΉςΈΣΨ≠ΒδΒΡΡΎΖ÷ΟΎΦΛΥΊΓΔ≈‘Ζ÷ΟΎΒςΫΎΦΝΜρ…ώΨ≠Βί÷ ΓΘ…ζ≥Λ“÷ΥΊ”–ΝΫ÷÷≤ΜΆ§ΒΡΜν–‘–Έ ΫΘΚ…ζ≥Λ“÷ΥΊ-14Θ®SST-14Θ©ΚΆ…ζ≥Λ“÷ΥΊ-28Θ®SST-28Θ©ΓΘSST-14 ΚΆ SST-28 ΨυΆ®ΙΐΗΏ«ΉΚΆΝΠΒΡΡΛ ήΧεΘ®…ζ≥Λ“÷ΥΊ ήΧε 1-5ΘΜSSTR1-5Θ©ΖΔΜ”…ζΈοΜν–‘Θ§’β–© ήΧεΙψΖΚΖ÷≤Φ”Ύ»ΥΧεΒΡΗς÷÷Ήι÷·÷–Θ§»γ…ώΨ≠Ήι÷·ΓΔ¥ΙΧεΓΔ…ω‘ύΓΔΖΈΚΆΟβ“ΏœΗΑϊΓΘ
…ζ≥Λ“÷ΥΊ ήΧεΘ®SSTRsΘ©‘ΎΕύ÷÷…ώΨ≠ΡΎΖ÷ΟΎΕώ–‘÷ΉΝωΘ®NETsΘ©÷–ΙΐΕ»±μ¥οΘ§Αϋά®“»œΌΓΔ¥ΙΧεΓΔ«ΑΝ–œΌΓΔΖΈάύΑ©ΓΔΙ«»βΝωΒ»Θ§“‘ΦΑ“Μ–©Ζ«…ώΨ≠ΡΎΖ÷ΟΎ÷ΉΝωΘ§»γ»ιœΌΑ©ΓΔΫα÷±≥ΠΑ©ΓΔ¬―≥≤Α©ΓΔΙ§Ψ±Α©Β»ΓΘ“ρ¥ΥΘ§’β–© ήΧεΩ…ΉςΈΣΑ–ΒψΘ§”Ο”ΎΫΪΗΏ–ß≈®Ε»ΒΡœΗΑϊΕΨ–‘“©Έο―Γ‘ώ–‘ΒΊΒίΥΆ÷Ν÷ΉΝω≤ΩΈΜΓΘ»ΜΕχΘ§Χλ»Μ…ζ≥Λ“÷ΥΊΜα“ρΟΗ¥ΌΫΒΫβΕχ―ΗΥΌΥ°ΫβΘ§“ρ¥ΥΩΣΖΔ≥ωΝΥΗϋΈ»Ε®«“–ßΝΠΗϋ«ΩΒΡάύΥΤΈοΓΘ’β–©άύΥΤΈοΆ®ΙΐΫΪ L-Α±ΜυΥαΧφΜΜΈΣΤδ D-“λΙΙΧεΘ§≤Δ±ΘΝτΫωΗΚ‘π…ζΈοΜν–‘ΒΡκΡ±μΈΜά¥ΥθΕΧκΡΝ¥ΕχΚœ≥…ΓΘΉνΙψΈΣ»Υ÷ΣΒΡ…ζ≥Λ“÷ΥΊάύΥΤΈο «ΜΖκΡΘ§Αϋά®Α¬«ζκΡΘ®d-Phe-c[Cys-Phe-d-Trp-Lys-Thr-Cys]-Thr-olΘ©ΓΔάΦ»πκΡΘ®d-2Nal-c[Cys-Tyr-d-Trp-Lys-Val-Cys]-Thr-NH2Θ©ΚΆΖΞΤ’κΡΘ®d-Phe-c[Cys-Tyr-d-Trp-Lys-Val-Cys]-Trp-NH2Θ©Θ§ΥϋΟ«÷ς“Σ”κ 2 –Ά ήΧεΘ®SSTR2Θ©ΫαΚœΘ§Εχ SSTR2 «“―÷ΣΙΐΕ»±μ¥οΉνΤΒΖ±ΒΡ…ζ≥Λ“÷ΥΊ ήΧεΓΘ”–ΕύΗω PDC ΒΡ Βάΐ”……œ ω…ζ≥Λ“÷ΥΊΑ–œρκΡΉι≥…Θ§ΜΙ”–ΤδΥϊ…ζ≥Λ“÷ΥΊκΡάύΥΤΈοΘ§άΐ»γΈεκΡ…ζ≥Λ“÷ΥΊΘ®DTPA-d-Phe-c[Cys-Phe-d-Trp-Lys-Thr-Cys]-Thr-olΘ©
±μΤΛ…ζ≥Λ“ρΉ”Θ®EGFΘ©
±μΤΛ…ζ≥Λ“ρΉ” ήΧεΘ®EGFRΘ© «“Μ÷÷ΩγΡΛΒΑΑΉΘ§ τ”Ύ ErbB Φ“ΉεΒΡ ήΧεά“Α±ΥαΦΛΟΗΘ§ΗΟΦ“Ήε”… 4 ÷÷ΫαΙΙœύΙΊΒΡ≥…‘±Ήι≥…ΘΚEGFR/HER1Θ®ErbB-1Θ©ΓΔHER2/neuΘ®ErbB-2Θ©ΓΔHER3Θ®ErbB-3Θ©ΚΆ HER4Θ®ErbB-4Θ©ΓΘΆ®Ιΐ…Η―Γ …ΨζΧε’Ι ΨΈΡΩβΘ§“―ΖΔœ÷–μΕύκΡΡή“‘ΗΏ«ΉΚΆΝΠΚΆ―Γ‘ώ–‘”κ EGFR ΫαΚœΘ§≤Δ“―±Μ”ΟΉςΑ–œρ“©ΈοΒίΥΆΒΡΩ…––ΖΫΖ®ΘΚYHWYGYT-PQNVIΓΔCMYIEALDKYACΓΔLTVSPWYΓΔYWPSVTLΓΘ
Angiopep-2ΘΚΉνΫϋ“ΐΤπΙΊΉΔΒΡ“Μ÷÷κΡ «“ΜΗωΟϊΈΣAngiopep-2Θ®TFFYGGSRGKRNNFK-TEEYΘ©ΒΡ 19 κΡΘ§“ρΤδΡήΙΜ¥©‘Ϋ―ΣΡ‘ΤΝ’œΘ®BBBΘ©Εχ±Η ή÷θΡΩΓΘ―ΣΡ‘ΤΝ’œ”…Ρ‘ΡΎΤΛœΗΑϊΙΙ≥…Θ§œό÷Τ≤ΔΩΊ÷ΤΉ≈÷– ύ…ώΨ≠œΒΆ≥”κ…μΧεΤδΥϊ≤ΩΈΜ÷°ΦδΒΡΖ÷Ή”ΫΜΜΜΓΘAngiopep-2ΡήΙΜΆ®Ιΐ”κ‘ΎΡ‘œΗΑϊ÷–Ιΐ±μ¥οΒΡΒΆΟήΕ»÷§ΒΑΑΉ ήΧεœύΙΊΒΑΑΉ -1Θ®LRP-1Θ©ΫαΚœΘ§Ψ≠ ήΧεΫιΒΦΒΡΉΣΑϊΆΧΉς”Ο¥©‘Ϋ―ΣΡ‘ΤΝ’œΓΘ¥ΥΆβΘ§Τδ–ρΝ–÷–Κ§”–ΒΡΝΫΗωάΒΑ±Υα ΙΤδ≥…ΈΣ“Μ÷÷”–Έϋ“ΐΝΠΒΡ PDC Κρ―ΓΈοΘ§÷Φ‘ΎΫΪ÷ΈΝΤ“©ΈοΆΒ‘Υ÷ΝΡ‘≤ΩΕώ–‘÷ΉΝωΓΘ
’κΕ‘…œΈΡΥυ ωΒΡ RGD κΡΜυ–ρΘ§“―ΩΣΖΔ≥ωΜΖκΡ±δΧεΓΘΉν≥Θ”ΟΒΡΜΖκΡ « iRGDΘ®CRGDKGPDCΘ©Θ§’β «“Μ÷÷ 9 ΗωΑ±ΜυΥαΒΡΜΖκΡΘ§ΨΏ”–¥©ΆΗ÷ΉΝωΉι÷·ΒΡΜν–‘ΓΘiRGD ≥θ Φ”κ÷ΉΝωΡΎΤΛœΗΑϊ÷–Ιΐ±μ¥οΒΡΠΝVΠ¬3 ΚΆΠΝVΠ¬5 ’ϊΚœΥΊΫαΚœΓΘΥφΚσΖΔ…ζΒΑΑΉΥ°Ϋβ«–ΗνΘ§±©¬Ε≥ωΈΜ”Ύ C ΕΥΒΡ“ΰΡδ RXXK/R Μυ–ρΘ®CendR Μυ–ρΘ§C ΕΥΙφ‘ρΘ©Θ§ΗΟΜυ–ρΥφΚσ”κ…ώΨ≠œΥΟΪΒΑΑΉ-1Θ®NRP-1Θ©ΫαΚœΘ§ΦΛΜνΡΎΆΧΉΣ‘ΥΆΨΨΕΘ§¥”Εχ‘ω«ΩΩΙΑ©“©Έοœρ÷ΉΝωΒΡ‘Υ δΘ®ΆΦ 2Θ©
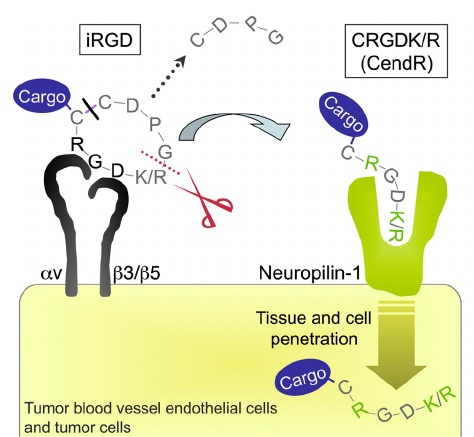
ΆΦ2ΘΚiRGD ΒΡΫαΚœ”κ…χΆΗΜζ÷ΤΓΘiRGD κΡ‘ΎΕώ–‘÷ΉΝω÷–±μ¥οΠΝv ’ϊΚœΥΊΒΡΡΎΤΛœΗΑϊΚΆΤδΥϊœΗΑϊ±μΟφΨέΦ·ΓΘRGD Μυ–ρΗΚ‘π”κ’ϊΚœΥΊΫαΚœΓΘΥφΚσΘ§ΗΟκΡ±ΜœΗΑϊ±μΟφœύΙΊΒΑΑΉΟΗ«–ΗνΘ§Ήν÷’‘Ύ C ΕΥΘ®Κλ…Ϊ–ιœΏΘ©±©¬Ε“ΰΡδΒΡ CendR ‘ΣΦΰ RXXK/RΓΘCendR ‘ΣΦΰΥφΚσΗ…»≈”κ…ώΨ≠œΥΟΪΒΑΑΉ -1 ΒΡΫαΚœΘ§¥”Εχ Βœ÷Ήι÷·ΚΆœΗΑϊ…χΆΗΓΘ÷ΉΝω…χΆΗκΡΩ…”Ο”Ύ–ό ΈΜθΈοΘ®ΦρΒΞΒΡΜ·―ßΜυΆ≈ΜρΡ…ΟΉΩ≈ΝΘΘ©Θ§ΒΪ«ΑΧα «ΜθΈο±Ί–κΝ§Ϋ”‘Ύ iRGD κΡΒΡ N ΕΥΘ§“ρΈΣκΡ‘ΎΡΎΜ·«ΑΕΰΝρΦϋ“―±Μ«–ΕœΘ®ΚΎ…ΪœΏΘ©
―Γ‘ώΚœ ΒΡ≈ΦΝΣ“©Έο
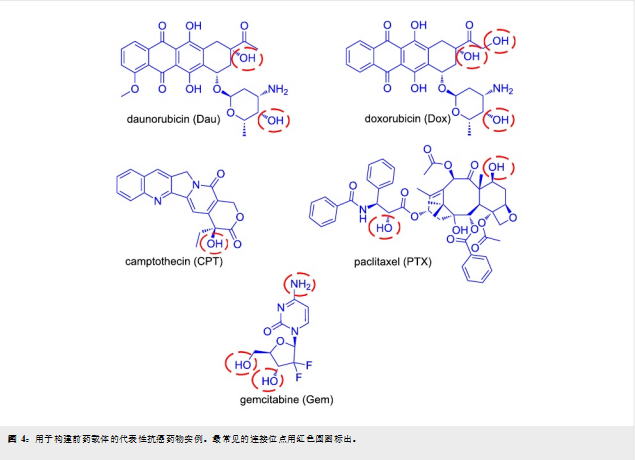
ΡΩ«Α”–Εύ÷÷“©Έο“―±Μ”ΟΉς PDC ÷–ΒΡΕΨ–‘Β·ΆΖΘ§Τδ÷–ΈεΗωΨΏ”–¥ζ±μ–‘ΒΡάΐΉ” «ΦΣΈςΥϊ±θΓΔΑΔΟΙΥΊΓΔ»αΚλΟΙΥΊΓΔΉœ…Φ¥ΦΚΆœ≤ ςΦνΘ®ΆΦ3Θ©ΓΘ’β–©‘≠ ΦΩΙΑ©“©ΈοΒΡ÷ς“Σ»±Βψ «ΤδΕΨ–‘≤Μ ήΩΊ÷ΤΘ§¥”ΕχΒΦ÷¬―œ÷ΊΒΡΗ±Ής”ΟΓΘ»γΙϊ≤ΜΧμΦ”Α–œρ≤ΩΖ÷Θ§ΥϋΟ««χΖ÷Α©œΗΑϊΚΆ’ΐ≥ΘœΗΑϊΒΡΡήΝΠΫœΒΆΓΘ¥ΥΆβΘ§ΧμΦ”κΡΉςΈΣΑ–œρ‘ΊΧεΩ…“‘ΧαΗΏ“©ΈοΒΡ“©¥ζΕ·ΝΠ―ßΚΆ÷ΈΝΤ¥ΑΩΎΓΘ”…”Ύ≤ΜΆ§ΒΡ“©ΈοΩ…Ρή≤…”Ο≤ΜΆ§ΒΡΜζ÷Τά¥…±ΥάœΗΑϊΘ§“ρ¥ΥΜαΗυΨίΑ–œρΑ©œΗΑϊΒΡΧΊ’ςά¥―Γ‘ώΚœ ΒΡ“©ΈοΓΘάΐ»γΘ§»αΚλΟΙΥΊΚΆΑΔΟΙΥΊΒΡΉς”ΟΜζ÷ΤœύΥΤΘ§ΕχΦΣΈςΥϊ±θΓΔœ≤ ςΦνΚΆΉœ…Φ¥ΦΒΡΉς”ΟΜζ÷Τ‘ρΗς≤ΜœύΆ§ΓΘ
Υυ―Γ“©Έο±Ί–κΖϊΚœΡ≥–©…ηΦΤ‘≠‘ρΘ§≤≈Ρή≥…ΈΣ PDC ΒΡάμœκΚρ―Γ“©ΈοΓΘΥυ―Γ“©Έο±Ί–κ ΚœΝ§Ϋ”Μ·―ßΖ¥”ΠΓΘΥϋ±Ί–κΨΏ”–Ω…÷±Ϋ””κκΡ/Ν§Ϋ”Ή”Ι≤ινΒΡΙΧ”–ΙΌΡήΆ≈Θ®ΆΦ3Θ©Θ§Μρ’ΏΨΏ”–ΡήΙΜ―ή…ζΜ·“‘Ϋχ––Ϋχ“Μ≤ΫΙ≤ινΒΡΙΌΡήΆ≈Θ®άΐ»γΘ§ΒψΜςΜ·―ßΘ©ΓΘ‘ΎΚσ“Μ÷÷«ιΩωœ¬Θ§±Ί–κΫς…ς―Γ‘ώ―ή…ζΜ·ΒΡΈΜ÷ΟΘ§“‘ΟβΗ…»≈“©ΈοΒΡ…ζΈοΜν–‘ΚΆΜν–‘“©ΈοΒΡ ΆΖ≈ΓΘ»γΙϊ“©ΈοΆ®Ιΐ Ε±πΧΊΕ® ήΧεΫχ––ΫαΚœΘ§‘ρ±Ί–κ≤…”ΟΦΤΥψΜζΡΘΡβΖΫΖ®ά¥Κœάμ―Γ‘ώΫΪΫχ––Μ·―ß–ό ΈΒΡ“©ΈοΈΜ÷ΟΓΘ
¥ΥΆβΘ§Υυ―Γ“©Έο±Ί–κΕ‘―ΓΕ®ΒΡΕώ–‘÷ΉΝωœΗΑϊΨΏ”–ΉψΙΜΒΡœΗΑϊΕΨ–‘Θ§“‘œϊ≥ΐΥϋΟ«≤ΔΫχΕχ¥ΌΫχ÷ΉΝωœϊΆΥΓΘάμœκ«ιΩωœ¬Θ§Υυ―Γ“©ΈοΕ‘ΡΩ±ξΕώ–‘÷ΉΝωΒΡΑκ ΐ“÷÷Τ≈®Ε»Θ®IC50Θ©÷Β”ΠΈΣΒΆΡ…ΡΠΕϊΦΕΓΘΩΥΖΰ“©Έο–ßΝΠΒΆΒΡ“ΜΗωΚœάμ≤Ώ¬‘ «‘ωΦ”κΡ‘ΊΧεΒΡ“©ΈοΗΚ‘ΊΝΩΓΘάΐ»γΘ§‘Ύ PDC ANG1005 ÷–Θ§ΒΞΗω―ΣΙήκΡ -2 κΡ…œΗΚ‘ΊΝΥ 3 Ηω“©ΈοΖ÷Ή”Θ®Ήœ…Φ¥ΦΘ©Θ§ΗΟ“©Έο“―Άξ≥… II ΤΎΝΌ¥≤ ‘―ιΓΘ»ΜΕχΘ§”κΆ®≥ΘΗϋ ή«ύμυΒΡΒΞ“©ΗΚ‘Ίœύ±»Θ§‘ωΦ”“©ΈοΗΚ‘ΊΒΡΗ≈ΡνΡ―“‘ Βœ÷Θ§’β÷ς“Σ «”…”ΎΤδΈοάμΜ·―ß–‘÷ ≤ΜΦ―ΓΘ
PDCΒΡΝ§Ϋ”Τς…ηΦΤ
‘Ύ…ηΦΤκΡ“©Έο≈ΦΝΣΈοΘ®PDCΘ© ±Θ§Νμ“ΜΗω–η“ΣΩΦ¬«ΒΡΙΊΦϋΖΫΟφ «Ν§Ϋ”κΡΚΆ“©ΈοΒΡΝ§Ϋ”Ή”ΓΘΝ§Ϋ”Ή”ΒΡΫαΙΙ±Ί–κΨΪ–Ρ…ηΦΤΘ§“‘Οβ”ΑœλκΡ”κΤδ ήΧεΒΡΫαΚœ«ΉΚΆΝΠ“‘ΦΑ“©ΈοΒΡΝΤ–ßΓΘ≤Μ«ΓΒ±ΒΡΝ§Ϋ”Ή”Ω…ΡήΜαΉηΑ≠“©Έο¥” PDC ÷– ΆΖ≈Θ§¥”ΕχΫΒΒΆΤδ’ϊΧε÷ΈΝΤ–ßΙϊΓΘ”Ο”Ύ PDC ΒΡΝ§Ϋ”Ή”÷÷άύΖ±ΕύΘ§ΥϋΟ«‘Ύ≥ΛΕ»ΓΔΈ»Ε®–‘ΓΔ ΆΖ≈Μζ÷ΤΓΔΙΌΡήΆ≈ΓΔ«ΉΥ°–‘/ ηΥ°–‘Β»ΖΫΟφΗς≤ΜœύΆ§ΓΘ
’β÷÷Ν§Ϋ”Ή”Ω…“‘…ηΦΤ≥…¥χ”–ΟΗΩ…Υ°ΫβΒΞ‘ΣΘ®EHUΘ©Θ§άΐ»γτ»ΥαθΞΦϋΜρθΘΑΖΦϋΘ§Ζ÷±πΩ…±ΜθΞΟΗΚΆθΘΑΖΟΗΥ°ΫβΓΘΉν≥Θ”Ο”ΎΝ§Ϋ”Ή”÷–¥χ”–τ»ΥαθΞΦϋΉςΈΣΟΗΥ°ΫβΒΞ‘ΣΒΡ «γζγξθΘΜυΘ®‘¥Ή‘γζγξΥαΘ©ΚΆΈλΕΰθΘΜυΘ®‘¥Ή‘ΈλΕΰΥαΘ©ΓΘΙΊ”Ύ‘ΎΝ§Ϋ”Ή”÷– Ι”ΟθΘΑΖΦϋΉςΈΣΝ§Ϋ”“©ΈοΚΆκΡΒΡΒΞ‘ΣΘ§Ω…“‘ΗυΨίΡΩ±ξΉι÷·ΚΆ/ΜρΑ©÷Δάύ–ΆΫχ––Ε®÷ΤΘ§“‘ ΙΤδ‘ΎΧΊΕ®ΒΡΒΑΑΉΟΗΘ®»γ‘ΎΑϋά®ΖΈΑ©ΓΔΡ‘Α©ΓΔ«ΑΝ–œΌΑ©ΚΆ»ιœΌΑ©‘ΎΡΎΒΡΕύ÷÷Εώ–‘÷ΉΝω÷–…œΒςΒΡΉι÷·ΒΑΑΉΟΗ BΘ©…œΒςΒΡ≤ΩΈΜ±ΜΥ°ΫβΓΘ¥ΥΆβΘ§‘Ύ…ηΦΤ PDC ±Θ§±Ί–κΧΊ±πΉΔ“β―Γ‘ώΝ§Ϋ”Ή”÷– Ι”ΟΒΡΦϋΓΘΨΏΧεΕχ―‘Θ§‘ΎΡΩ«ΑΩ…”ΟΒΡ–μΕύ PDC ÷–Θ§÷Ν…Ό Ι”ΟΝΥΝΫ÷÷≤ΜΆ§ΒΡΦϋΘΚ“Μ÷÷”Ο”ΎΫΪΝ§Ϋ”Ή”Ν§Ϋ”ΒΫκΡ…œΘ§Νμ“Μ÷÷”Ο”ΎΫΪ“©ΈοΝ§Ϋ”ΒΫΝ§Ϋ”Ή”…œΓΘ‘Ύ…ηΦΤΙΐ≥Χ÷–Θ§¥Υάύ«ιΩω±Ί–κΩΦ¬«ΉιΉΑΚΟΒΡ«Α“©‘ΊΧεΘ®PDCΘ©Υυ¥ΠΒΡΈΔΜΖΨ≥Θ§“ρΈΣ≤ΜΆ§ΒΡΟΗΚΆ/Μρ÷ΉΝωΈΔΜΖΨ≥Ω…ΡήΜαΒΦ÷¬“©Έο¥” PDC ÷–Ιΐ‘γ ΆΖ≈Θ§Φ¥Ήν÷’ ΆΖ≈≥ω–·¥χ“©ΈοΒΡΝ§Ϋ”≤ΩΖ÷ΓΘ
Νμ“ΜάύΝ§Ϋ”Χε «¥ΧΦΛœλ”Π/Ω…ΫΒΫβΝ§Ϋ”ΧεΘ§÷Φ‘Ύ Βœ÷“©Έο¥”…ζΈο≈ΦΝΣΈο‘Ύ÷ΉΝωΈΔΜΖΨ≥÷–ΒΡΗΏ–ß ΆΖ≈ΓΘ’βάύΝ§Ϋ”ΧεΨ≠ΙΐΚœάμ…ηΦΤΘ§ΡήΙΜ‘ΎΑ©œΗΑϊΜΖΨ≥Θ®ΈΔΥα–‘ pH ÷ΒΓΔΜΙ‘≠ΦΝΚΆ/ΜρΟΗΥ°ΤΫ…ΐΗΏΘ©ΜρΆβ≤Ω¥ΧΦΛΘ®≥§…υ≤®ΓΔΈ¬Ε»ΓΔΖχ…δΘ©œ¬Η–÷ΣΧΊΕ®¥ΧΦΛ ±±Μ«–ΕœΓΘΨΏΧεΕχ―‘Θ§Ρ≥–©Φϋ»γ―«ΑΖΦϋΓΔκΩΦϋΓΔκξΦϋΓΔΝΎθΞΦϋΓΔΥθ»©ΦϋΓΔ““œ©ΜυΟ―ΦϋΚΆΨέΥθ»©ΦϋΘ§‘ΎΥα–‘ pH ÷ΒΧθΦΰœ¬ΜαΖΔ…ζΥ°ΫβΘ§Εχ‘Ύ―Σ“Κ―≠ΜΖΙΐ≥Χ÷–»¥ΦΪΤδΈ»Ε®ΓΘ“ρ¥ΥΘ§ΥαΟτΗ–ΦϋΡήΙΜ‘ΎΑ©œΗΑϊΒΡΈΔΥα–‘ΈΔΜΖΨ≥ΚΆ/ΜρœΗΑϊΡΎΥα–‘«χ “÷–Υ°ΫβΘ§¥”Εχ ΆΖ≈≥ωΜν–‘“©ΈοΓΘ¥ΥΆβΘ§ΕΰΝρΦϋ‘ΎΩΙΧε“©Έο≈ΦΝΣΈο÷–“≤≥Θ±Μ≤…”ΟΘ§“ρΈΣΥϋΟ«Μα±ΜΕώ–‘œΗΑϊ÷–ΗΏ≈®Ε»¥φ‘ΎΒΡΑκκΉΑ±ΥαΚΆΙ»κΉΗ κΡΒ»ΜΙ‘≠ΦΝ«–ΕœΓΘ
¥χ”–ΟΗΥ°ΫβΒΞ‘ΣΘ®EHUΘ©«“Ε‘ΒΑΑΉΟΗ”–œλ”ΠΒΡΝ§Ϋ”Ή” «Ω…ΫΒΫβΒΡκΡΝ§Ϋ”Ή”Θ§”…”ΎΡ≥–©ΟΗΒΡΧΊ“λ–‘Θ§’βάύΝ§Ϋ”Ή”“ΐΤπΝΥ»ΥΟ«ΒΡΦΪ¥σΙΊΉΔΘ§ΫϋΡξά¥ΤδΖΔ’Ι ΤΆΖ―ΗΟΆΓΘΗΟΝλ”ρΉνΨΏ¥ζ±μ–‘ΒΡάΐΉ” «Μυ÷ Ϋπ τΒΑΑΉΟΗΘ®MMP-2/9Θ©ΚΆΉι÷·ΒΑΑΉΟΗ B ΒΡκΡΒΉΈοΓΘMMP-2/9 ΚΆΉι÷·ΒΑΑΉΟΗ B «‘ΎΑ©œΗΑϊ÷–Κ§ΝΩΫœΗΏΒΡΒΑΑΉΥ°ΫβΟΗΘ§≤Έ”κ»Υάύ÷ΉΝωΒΡ«÷œ°ΚΆΉΣ“ΤΓΘΉι÷·ΒΑΑΉΟΗ B ΡήΙΜ Ε±πΧΊΕ®ΒΡκΡ–ρΝ–Θ§»γ Val-CitΘ®γ”Α±Υα-ΙœΑ±ΥαΚΆGFLGΓΘΝμ“ΜΖΫΟφΘ§GPLGIAGQΓΔPLGLAGΚΆ GPVGLIGK «Μυ÷ Ϋπ τΒΑΑΉΟΗ -2 ΚΆΜυ÷ Ϋπ τΒΑΑΉΟΗ -9 ΒΡ“Μ–©≥ΘΦϊκΡΒΉΈοΓΘ
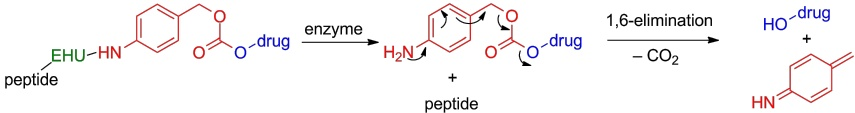
ΆΦ4ΘΚΆ®ΙΐΟΗΥ°ΫβΒΞ‘ΣΫΪ“©Έο ΆΖ≈Μζ÷Τ¥”Ή‘ΜΌΦδΗτΈο PABC ”κ÷ΉΝωΙι≥≤κΡΝ§Ϋ”Τπά¥ΒΡΆΦ ΨΓΘΚλ…Ϊ = Ή‘ΜΌΦδΗτΈο PABCΘΜάΕ…Ϊ = “©ΈοΘΜ¬Χ…Ϊ = ΟΗΥ°ΫβΒΞ‘ΣΘ®EHUΘ©ΘΜΚΎ…Ϊ = ÷ΉΝωΙι≥≤κΡ
‘ΎΙΐ»ΞΒΡΦΗΡξ÷–Θ§Ψέ““Εΰ¥ΦΜ·“©Έο≈ΦΝΣΈοΘ®PDCΘ©Ν§Ϋ”Ή”÷–Νμ“ΜΗω―ΗΥΌ–ΥΤπ«“±Η ήΙΊΉΔΒΡάύ±π «Ή‘œϊΫβΜρΉ‘ΜΌ–‘ΦδΗτΜυ/Ν§Ϋ”Ή”ΓΘ’βάύΦδΗτΜυ/Ν§Ϋ”Ή”ΡήΙΜΆ®ΙΐΦΕΝΣΖ¥”ΠΆ§ ± ΆΖ≈Μν–‘“©ΈοΘ§»γΆΦ4Υυ ΨΓΘΕ‘Α±Μυή–¥ΦΘ®PABCΘΜΚλ…Ϊ≤ΩΖ÷Θ©ΨΆ «“ΜΗωΒδ–ΆΒΡάΐΉ”Θ§ΤδΑ±ΜυΩ…“‘Ά®ΙΐθΘΑΖΦϋ”κΟΗΩ…Υ°ΫβΒΞ‘ΣΘ®EHUΘΜ¬Χ…Ϊ≤ΩΖ÷Θ©œύΝ§Θ§Ά§ ±”κ÷ΉΝωΑ–œρ‘ΣΦΰΘ®»γ÷ΉΝωΙι≥≤κΡΘΜΚΎ…Ϊ≤ΩΖ÷Θ©œύΝ§ΓΘΤδΝμ“ΜΕΥΒΡτ«ΜυΩ…“‘Ά®ΙΐΧΦΥαθΞ/Α±ΜυΦΉΥαθΞΦϋ”κœΗΑϊΕΨ–‘“©ΈοΘ®άΕ…Ϊ≤ΩΖ÷Θ©œύΝ§ΓΘEHU ±Μ…ηΦΤ≥…Α–œρ÷ΉΝωΈΔΜΖΨ≥÷–Ιΐ±μ¥οΒΡΒΑΑΉΟΗΘ®»γΉι÷·ΒΑΑΉΟΗ BΘ©ΒΡΒΉΈοΓΘ“ΜΒ©±Μ’β–©ΟΗ Ε±πΘ§EHU ΨΆΜα±Μ«–Ηνœ¬ά¥Θ§¥”ΕχΆ®ΙΐΩλΥΌΦΕΝΣΖ¥”Π ΆΖ≈≥ωΜν–‘“©ΈοΘ®ΆΦ 4Θ©
≤ΈΩΦΈΡœΉΘΚBeilstein J. Org. Chem. 2018, 14, 930®C954.doi:10.3762/bjoc.14.80
